Research Group: Pulmonary Arterial Hypertension and Right Heart (Group Leader: Dr. Sarah Costantino)
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by elevated pulmonary arterial pressures and right ventricular hypertrophy. While treatments to delay disease progression are available, PAH has a poor prognosis eventually leading to right heart failure and death.
PAH is characterized by constant remodeling of the pulmonary artery by increased proliferation and migration of pulmonary arterial smooth muscle cells (PASMCs), fibroblasts (FBs) and endothelial cells (ECs). Such adverse remodeling eventually leads to increased pressure in the right ventricle with subsequent right ventricular hypertrophy and failure. Although inborn genetic variation plays an important role in PAH, several factors including drugs and environmental cues can significantly contribute to its pathogenesis. While much attention was drawn into investigating and developing therapies related to the most well understood signaling pathways in PAH, in the last decade, a shift towards understanding the gene-environment interaction driving the disease occurred. The Costantino lab investigates the transcriptional regulation, cell-cell interaction and phenotypic changes underpinning PAH development. These experimental and translational aims will be accomplished by state-of-the-art molecular biology, conditional mouse models and unique biobanks of PAH patients (Figure).
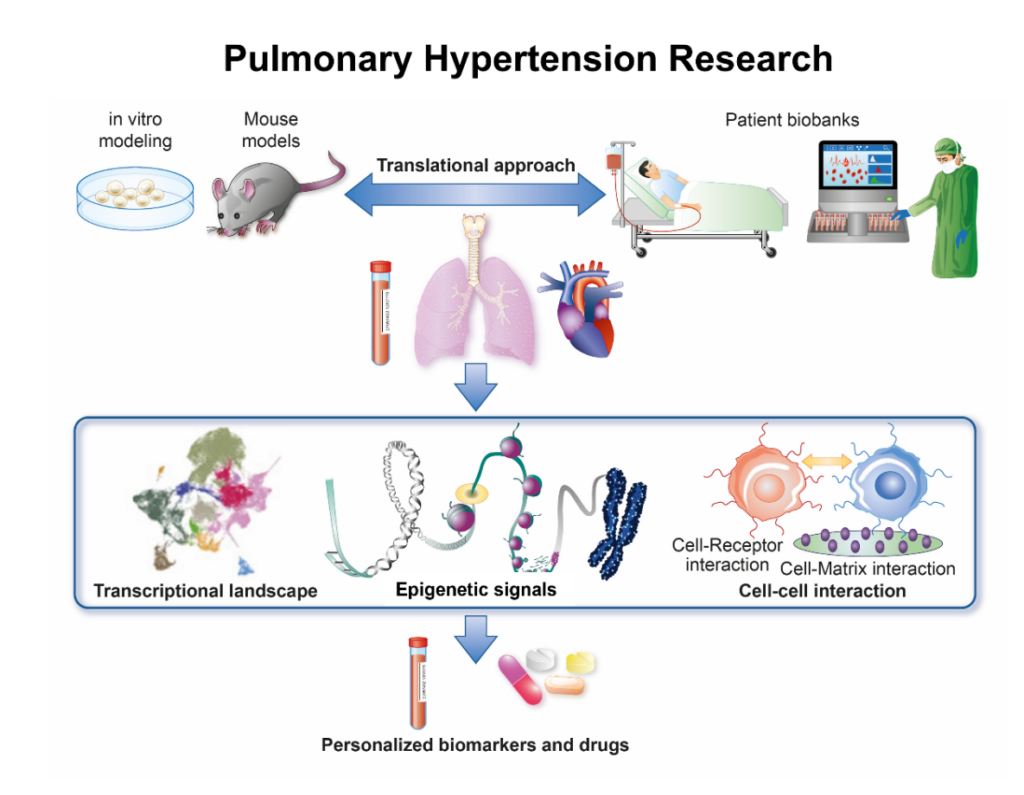 Figure. Translational approach to PAH
Figure. Translational approach to PAH
Specific research lines
- Characterize the mechanisms of microvascular endothelial dysfunction in PAH by leveraging a multi-omic approach
- Study the role of epigenetic signals (DNA methylation, histone modifications and non-coding RNAs) in orchestrating transcriptional programs underlying PAH phenotypes
- Investigate the cross-talk among different cells in experimental models of pre- and post-capillary PAH
- Develop novel PAH biomarkers
Research Team
- Alessandro Mengozzi, MD, PhD
- Marialucia Telesca (Post Doc)
- Jacopo Spezzini (PhD Student)
- Valeria Masciovecchio (Master Student)
Funding
- Swiss Heart Foundation
- Gebauer Stiftung
- Swiss Life Foundation
- Holcim Stiftung
- Mengozzi A*, Costantino S*, Paneni F, Duranti E, Nannipieri M, Mancini R, Lai M, La Rocca V, Puxeddu I, Antonioli L, Fornai M, Ghionzoli M, Georgiopoulos G, Ippolito C, Bernardini N, Ruschitzka F, Pugliese NR, Taddei S, Virdis A, Masi S. Targeting SIRT1 Rescues Age- and Obesity-Induced Microvascular Dysfunction in Ex Vivo Human Vessels. (*equally contribution) Circ Res. 2022. doi: 10.1161/CIRCRESAHA.122.320888.
- Ambrosini S, Montecucco F, Kolijn D, Pedicino D, Akhmedov A, MohammedSA, Herwig M, Gorica E, Lujza Szabó P, Weber L, Russo G, Vinci R, MatterCM, Liuzzo G, Brown PJ, Rossi FMV, Camici GG, Sciarretta S, Paolo BeltramiA, Crea F, Podesser B, Lüscher TF, Kiss A, Ruschitzka F, Hamdani N, Costantino S*, Paneni F*. Methylation of the Hippo effector YAP by themethyltransferase SETD7 drives myocardial ischemic injury: a translational study. Cardiovasc Res. 2022. doi: 10.1093/cvr/cvac102. PMID: 35709329 (*equally contribution)
- Masi S, Ambrosini S, Mohammed SA, Sciarretta S, Lüscher TF, Paneni F, Costantino S. Epigenetic Remodeling in Obesity-Related Vascular Disease. Antioxid Redox Signal. 2020; 34(15):1165-1199. PMID: 32808539
- Mohammed SA, Albiero M, Ambrosini S, Gorica E, Karsai G, Caravaggi CM, Masi S, Camici GG, Wenzl F, Calderone V, Madeddu P, Sciarretta S, Matter CM, Spinetti G, Luscher TF, Ruschitzka F, Costantino S*, Fadini GP*, Paneni F. The BET Protein Inhibitor Apabetalone Rescues Diabetes-induced Impairment of Angiogenic Response by Epigenetic Regulation of Thrombospondin-1. Antioxid Redox Signal. 2021. DOI:10.1089/ars.2021.0127. PMID: 34913726 (*equally contribution)
- Hamdani N, Costantino S, Mügge A, Lebeche D, Tschöpe C, Thum T, Paneni F. Leveraging clinical epigenetics in heart failure with preserved ejection fraction: a call for individualized therapies. Eur Heart J. 2021; 42(20):1940-1958. PMID: 33948637
- Costantino S, Akhmedov A, Melina G, Mohammed SA, Othman A, Ambrosini S, Wijnen WJ, Sada L, Ciavarella GM, Liberale L, Tanner FC, Matter CM, Hornemann T, Volpe M, Mechta-Grigoriou F, Camici GG, Sinatra R, Lüscher TF, Paneni F. Obesity-induced activation of JunD promotes myocardial lipid accumulation and metabolic cardiomyopathy. Eur Heart J. 2019; 40:997-1008. PMID: 30629164.
- Costantino S, Paneni F, Virdis A, Hussain S, Mohammed SA, Capretti G, Akhmedov A, Dalgaard K, Chiandotto S, Pospisilik JA, Jenuwein T, Giorgio M, Volpe M, Taddei S, Lüscher TF, Cosentino F. Interplay among H3K9-editing enzymes SUV39H1, JMJD2C and SRC-1 drives p66Shc transcription and vascular oxidative stress in obesity. Eur Heart J. 2019; 40:383-391. PMID: 29077881
- Costantino S, Libby P, Kishore R, Tardif JC, El-Osta A, Paneni F. Epigenetics and Precision Medicine in Cardiovascular Patients: From Basic Concepts to the Clinical Arena. Eur Heart J. 2018; 39:4150-4158. PMID: 29069341