(Neuro)endocrine Tumors
Untersuchung neuer personalisierter Therapieoptionen (molekular-gezielter Therapien, SSTR2-gesteuerter Radiorezeptortherapie), von Therapieresistenzen und deren Überwindung an neuroendokrinen Tumoren, Phäochromozytomen und Paragangliomen in vitro, in vivo und an humaner Primärkulturkultur.
AG Nölting / (Neuro)endokrine Tumore & PPGLs
Neuroendokrine Tumore (NET) sind eine heterogene Gruppe potentiell maligner Tumore. Sie sind hauptsächlich im gastroenteropankreatischen System, meist Dünndarm oder Bauchspeicheldrüse, sowie der Lunge lokalisiert. Sie gehen von den neuroendokrinen Zellen des gastroenteropankreatischen Systems bzw. der Lunge aus und können verschiedene Hormone (wie Insulin, Gastrin oder Serotonin) – je nach Entität – produzieren. Eine Besonderheit dieser Tumore ist ihre Überexpression des Somatostatinrezeptors 2 (SSTR2). Bei Erstdiagnose sind bereits 40-45% der Tumore metastasiert und benötigen daher eine systemische Therapie. Aktuell gibt es leider nur wenige systemische Therapieoptionen. Die einzig bis dato zugelassenen systemischen Therapieoptionen umfassen: SSTR2-Analoga (für die wachstumshemmende Indikation nur bei einem niedrigen Proliferationsindex Ki67 < 10% zugelassen), die SSTR2-gesteuerte Radiorezeptortherapie und die beiden molekular gezielten Therapien Everolimus (mTORC1 Inhibitor) und Sunitinib (Tyrosinkinaseinhibitor, nur für NET der Bauchspeicheldrüse zugelassen). Nicht offiziell in Europa zugelassen, aber in klinischem Gebrauch sind noch einzelne Chemotherapeutika.
Ähnliches gilt für die Adrenalin/Noradrenalin produzierenden Tumore des Nebennierenmarks (Phäochromozytome) und der sympathischen und parasympathischen Paraganglien (Paragangliome) des autonomen Nervensystems. Auch sie über-exprimieren häufig SSTR2. Es gibt bislang keine offiziell zugelassenen Therapieoptionen für Phäochromozytome/Paragangliome.
Somit erforschen wir an oben genannten neuroendokrinen Tumoren, Phäochromozytomen und Paragangliomen neue personalisierte molekular-gezielte Therapieoptionen sowie Resistenzentwicklung gegenüber bereits zugelassenen Medikamenten wie Everolimus und deren Überwindung in vitro, in vivo und an humaner Primärkultur. Ein weiteres Ziel unserer Forschung ist die Verbesserung der Wirksamkeit der SSTR2-gesteuerten Radiorezeptortherapie über Induktion der SSTR2-Rezeptoren.

Research Topics
Characterisation of the mechanisms of resistance to the mTORC1 inhibitor everolimus, and their management, in pancreatic neuroendocrine tumors (NET)
The mTORC1 inhibitor everolimus is one of the few officially approved therapies for NETs. However, after initial disease stabilisation, most patients develop resistance to everolimus after less than one year of therapy. We have established and characterised the first stable everolimus-resistant pancreatic NET cell line model (BON1 RR1/RR2), and demonstrated therapy to overcame such resistance in vitro, and transferred the resistant cell line model to an orthotopic intrapancreatic NET xenograft mouse model (Aristizabal Prada, et al. 2018). The aim of our current project is to further characterise the first everolimus-resistant orthotopic pancreatic NET xenograft mouse model, and investigate whether the PI3K inhibitor alpelisib – which overcame resistance in vitro – can also overcome resistance in vivo. If successful, this therapeutic approach could directly be translated to the clinic since alpelisib is already in clinical use (approved for breast cancer). Moreover, we plan to investigate potential novel markers of everolimus resistance in the mouse tumors, and explore and hopefully confirm these markers in human tumor tissue microarrays in order to implement novel markers of everolimus resistance for NET patients.
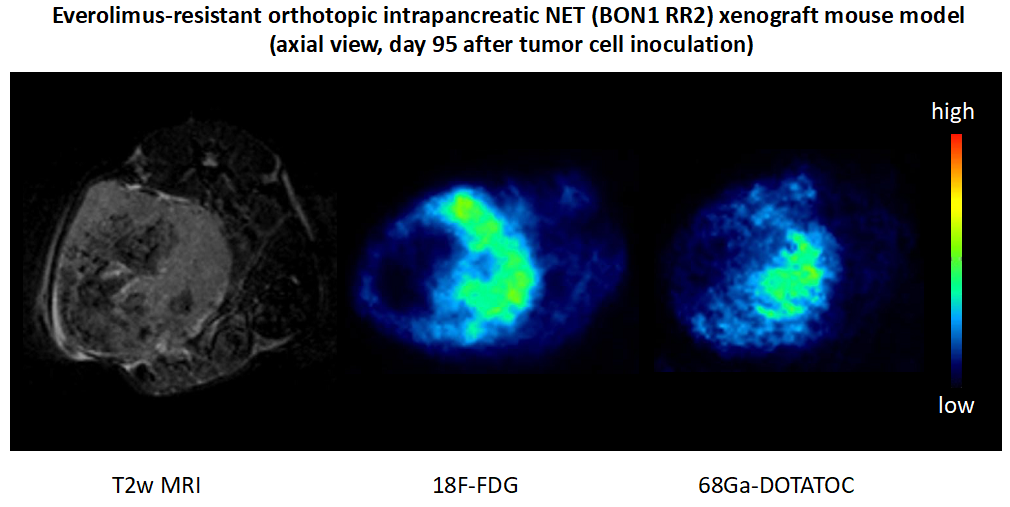
Therapy resistance in neuroendocrine neoplasia
Together with the German “NET-Z Werk”, a network of several German NET researchers, we have established three additional everolimus-resistant pancreatic NET cell lines (two from human primary cultures) with reversible resistance, and characterised them in detail. We then compared them to the irreversibly resistant BON1 RR1/RR2 cell lines (see above) by whole-exome sequencing, proteomics and metabolomics. This is now enabling us to decipher the mechanisms of reversible versus stable resistance to everolimus. Moreover, we have already generated human tumor tissue microarrays with samples from patients who responded to everolimus and patients who did not respond in order to establish potential markers of reversible versus irreversible resistance.
Radiosensitizing and SSTR re-expression in NET cells: Translational concepts for peptide receptor chemo-radionuclide therapy (PRCRT)
SSTR2-guided radionuclide therapy is an officially approved, effective, and well-tolerated systemic therapy option for NETs. It may also be a promising therapy option for other tumor entities with high SSTR2 expression, such as pheochromocytomas/paragangliomas and thyroid carcinomas, as well as non-endocrine tumors, for which it has not yet been officially approved. We have demonstrated that the combination of 5-fluorouracil with epigenetic modifiers induces radiosensitization, SSTR2 expression and radioligand binding in NET cells in vitro (Jin, et al. 2020). This so-called peptide receptor chemo-radionculide therapy (PRCRT) may help to further improve objective response rates and progression-free survival in NETs. We aim to further investigate and improve possible drug combinations for SSTR-based PRCRT and to expand our findings to non-neuroendocrine cancers. By increasing SSTR2 expression and radioligand binding of tumor cells on the one hand, and causing radiosensitization on the other hand, this approach might further improve SSTR-based PRCRT in NETs and might help to broaden the indication of SSTR-based PRCRT to various non-neuroendocrine cancer types.
The molecular pathology of pheochromocytomas / paragangliomas (PPGLs) and genetically-driven molecular-targeted therapies: Towards precision medicine
Each PPGL has the potential to spread to distant sites, and there are no officially approved systemic therapy options in Europe. Approximately 30-35% of PPGL patients show germline, and another 35-40% somatic driver-mutations. Thus, around 70% of all PPGLs can be assigned to one of three specific molecular clusters with different gene expression signatures and clinical behavior. The aim of this project is to implement personalised genetically-driven therapies for PPGLs (cluster-dependent therapies). We have established 2D/3D human PPGL primary cultures for multiple drug testing (Fankhauser, et al. 2019), and we have collected surgery-derived human primary cultures from more than 25 different PPGL patients. All surgery-derived tumors have been sequenced. Patients have been tested for germline mutations (if they agreed). The identified germline/somatic mutations were correlated with drug responsiveness (to more than 15 different drugs) and downstream pathway alterations of the derived primary cultures, as well as with histopathological and clinical data. We plan to collect primary cultures from approximately 60 different PPGL patients in total. The results will be cross-validated in different PCC cell line models, spheroids, in vivo allograft mouse models and patient-derived xenografts.
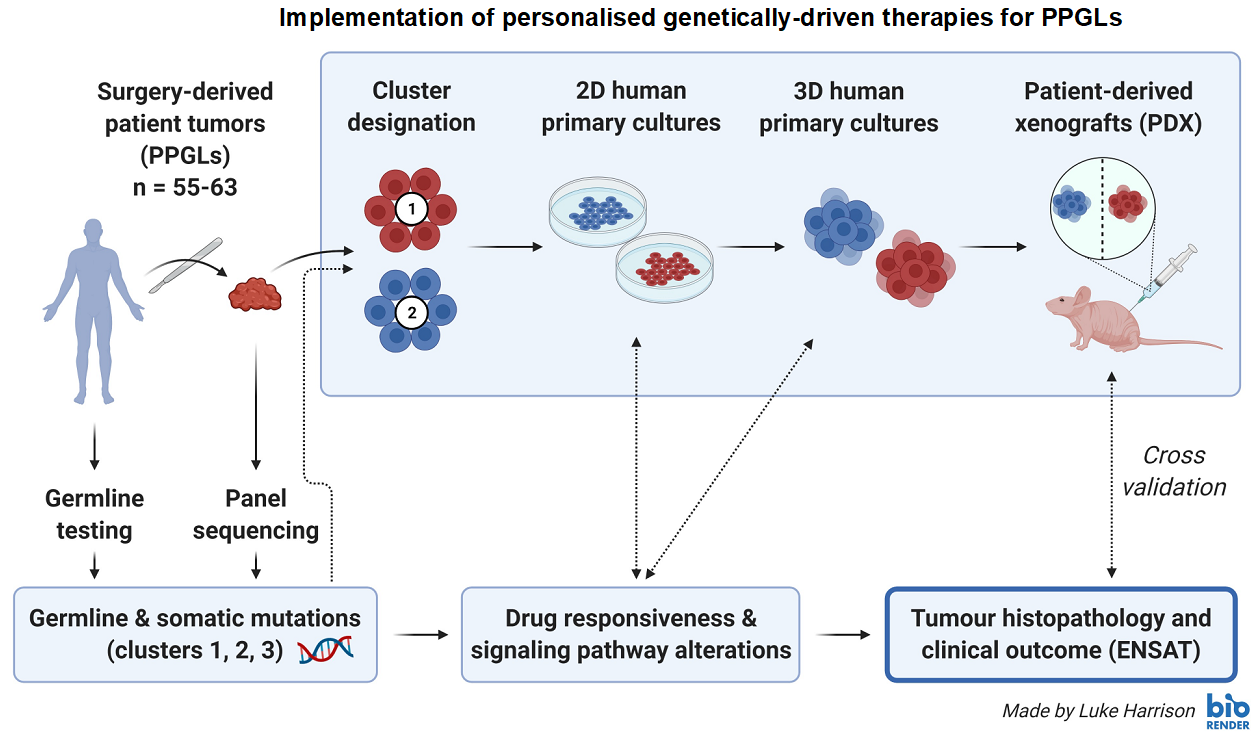
Cooperations
- Dr. Markus Manz, Department of Medical Oncology and Hematology, University Hospital of Zurich, Switzerland
- Sc Christian Pellegrino, Department of Medical Oncology and Hematology, University Hospital of Zurich, Switzerland
- Dr. Dario Neri, Institute of Pharmaceutical Sciences, Department of Chemistry and Applied Biosciences ETH Zürich, Switzerland
- PD Dr. Diana Vetter, Department of Surgery, University Hospital of Zurich, Switzerland
- Juliane Friemel, Institute of Pathology, University Hospital of Zurich, Switzerland
- PD Dr. Broglie Däppen, ORL, University Hospital of Zurich, Switzerland
- Dr. Ashley Grossman, Oxford Center for Diabetes, Endocrinology and Metabolism, University of Oxford, and Centre for Endocrinology, Barts and the London Scool of Medicine, University of London, UK
- Dr. Karel Pacak, NICHD, NIH, Bethesda, Maryland, US
- Dr. Christoph Auernhammer, Prof. Dr. Matthias Kroiss, Prof. Dr. Klaus Parhofer, Department of Medicine IV, University Hospital, LMU Munich, Germany
- PD Dr. Michael Lauseker, Institute for Medical Information Sciences, Biometry, and Epidemiology, LMU Munich, Germany
- Dr. Thomas Knösel, Institute of Pathology, LMU Munich, Germany
- PD Dr. Matthias Ilmer, PD Dr. Florian Bösch, Department of Surgery, University Hospital, LMU Munich, Germany
- Natalia Pellegata, Helmholtz Center Munich, Germany
- Prof Dr. Graeme Eisenhofer, Dr. rer nat. Nicole Bechmann , PD Dr. Christian Ziegler, University Hospital Carl Gustav Carus Dresden, Germany
- Dr. Jens Pietzsch, Dr. Martin Ullrich, Helmholtz Center Dresden, Germany
- Barbara Klink, Doreen William, Institute for Clinical Genetics, Faculty of Medicine Carl Gustav Carus TU Dresden, Germany
- Nicola Beindorff, Prof. Winfried Brenner, Department of Nuclear Medicine, University Hospital Charité Berlin, Germany
- PD Dr. Patricia Grabowski, Klinikum Bad Berka & University Hospital Charité Berlin, Germany
- PD Dr. Jörg Schrader, Department of Medicine I, University Hospital Hamburg-Eppendorf, Germany
- PD Dr. Sebastian Krug, Department of Internal Medicine I, University Hospital Halle-Wittenberg, Germany
- Dr. Martin Fassnacht, Department of Medicine I, University Hospital Würzburg, Germany
Fundings
German Research Foundation (DFG)
Else Kröner-Fresenius Foundation (EKFS)
HMZ Flagship Project Immuno-TargET
Novartis, Young Investigators in Neuroendocrine Neoplasia Germany Program (YING) European Funding (H2020)


