SSc, fibrosis and autoimmunity
Systemic sclerosis is a multisystem autoimmune fibrotic disease. We are using this disease as a model to understand the mechanisms of fibrosis in skin and lung disease. We are also interested in vasculopathy contributing to fibrosis in systemic sclerosis.
Our special interest is on non-coding RNA, including microRNA and long non-coding RNA. We are using 2D and 3D cultures, as well as animal models to understand and characterise mechanisms of fibrosis. Our overall aim is to develop new therapies for fibrotic diseases.
Current projects
Fibrosis is the most severe process in the pathophysiology of SSc. The tissue is replaced with a collagen rich, stiff connective tissue leading to dysfunction and failure of affected organs (1). Long non-coding RNAs (lncRNAs) are a recently emerging class of important gene regulators mediating their effects via a wide variety of mechanisms. LncRNAs are thought to be higher in numbers than coding RNAs, underlining their potential as master regulators of biological pathways and mediators of diseases (2). However, little is known about the role of lncRNAs in fibrotic diseases.
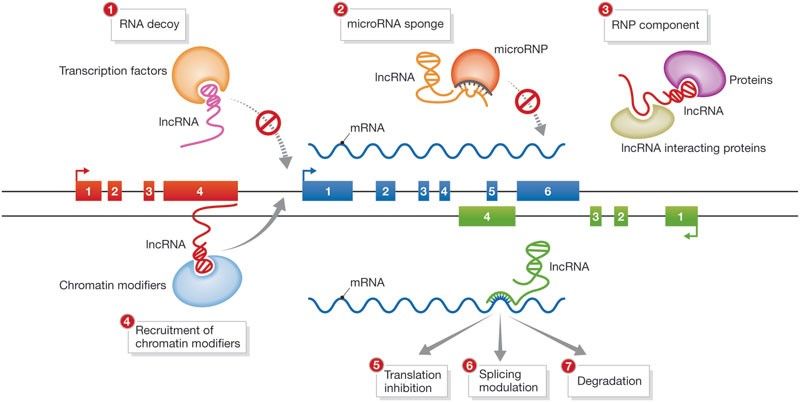
Hu W et al., EMBO reports, 2012
We hypothesize that lncRNAs might be key drivers in the pathophysiology of fibrosis in SSc. Therefore, lncRNA characterization might shed lights on novel fibrotic molecular mechanism. Here, we aim to identify uncharacterized lncRNAs using different screening technique like whole genome RNA sequencing or RT-qPCR on SSc vs healthy skin biopsies or fibroblasts. Furthermore, we aim to define the fibrotic molecular cascades that trigger lncRNAs dysregulation. In parallel, using gain and loss of function technologies and functional assay, we will address the role of the newly identified lncRNAs. New high-throughput, cutting-edge techniques like ATAC-seq, ChiRP or RNA pull down will be use to asses lncRNAs interactome and define the molecular mechanism of action (3,4).
References
- Gabrielli A, Avvedimento E V, Krieg T. Scleroderma. N Engl J Med. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved.; 2009 Jan;360(19):1989–2003.
- Cech TR, Steitz JA. The noncoding RNA revolution – Trashing old rules to forge new ones. Cell. 2014. p. 77–94.
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–8.
- Chu C, Spitale RC, Chang HY. Technologies to probe functions and mechanisms of long noncoding RNAs. Nat Struct Mol Biol. 2015;22(1):29–35.
Medications directly targeting fibrosis and fibrosis related mechanism are very limited, leaving a high-unmet need for rapid development of novel anti-fibrotic treatments. Skin fibrosis is an excellent surrogate to study the efficacy of anti-fibrotic drug candidates for SSc. Also, because of the overlapping pathophysiology with general fibrotic mechanisms, anti-fibrotic medications for SSc are top candidates for targeted therapies in other fibrotic diseases as well (1). The current concepts of preclinical drug development in SSc is focusing on two-dimensional cell cultures and animal models. Both models are useful, but have important limitations (2, 3). For this reason, we are developing a novel three-dimensional human microtissue skin model, where multiple cell types are included (fibroblasts, keratinocytes, endothelial cells and macrophages). Our model has the potential to booster preclinical drug development for fibrotic skin diseases, and to improve the predictability of clinical efficacy of anti-fibrotic drugs. Additionally, it has a high translational potential for fibrotic diseases in general.
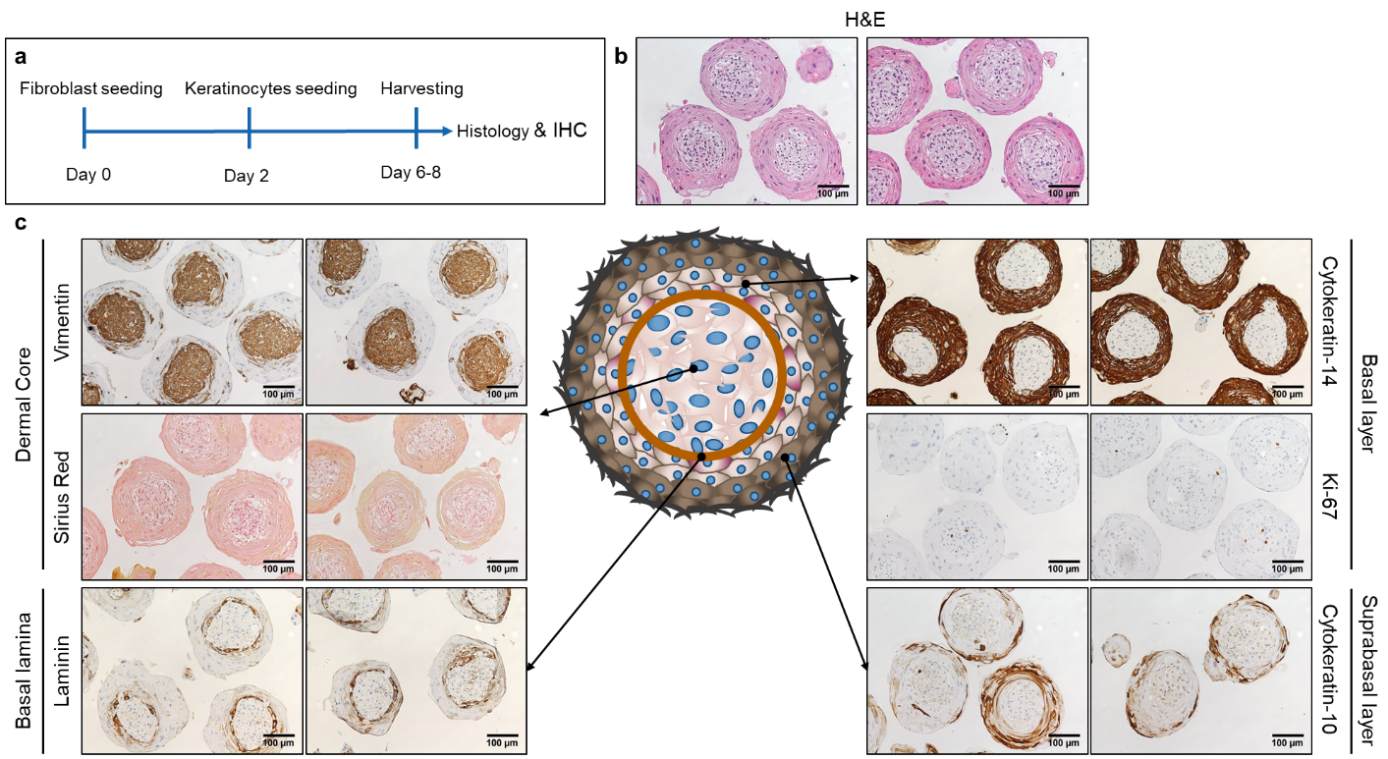
Schematic representation of the experimental setup for the microtissue production (a). Haematoxylin eosin staining (b). Immunohistochemistry staining for vimentin, laminin, cytokeratin 14, cytokeratin 10 and Ki-67 and Sirius Red staining (c).
References
- Palumbo-Zerr, K., et al., Orphan nuclear receptor NR4A1 regulates transforming growth factor-beta signaling and fibrosis. Nat Med, 2015. 21(2): p. 150-8.
- Pachera, E., et al., Long noncoding RNA H19X is a key mediator of TGF-beta-driven fibrosis. J Clin Invest, 2020. 130(9): p. 4888-4905.
- Mestas, J. and C.C. Hughes, Of mice and not men: differences between mouse and human immunology. J Immunol, 2004. 172(5): p. 2731-8.
We are performing high-throughput screening assay for tool compounds for SSc and fibrosis. A major hallmark of SSc fibroblasts is the differentiation into highly activated alpha smooth muscle actin (α-SMA) positive myofibroblasts that release large amounts of extracellular matrix components (1, 2). TGF-β was characterized as one of the main factors driving the activation of fibroblasts into myofibroblasts in the early active phase of the disease (3). Here, large compound libraries are tested on SSc fibroblasts for the ability to prevent myofibroblast differentiation using an automated platform. Immunofluorescence staining for the myofibroblast marker alpha smooth muscle actin (αSMA) is used as the endpoint. Characterization of the candidate anti-fibrotic compounds will be also performed.
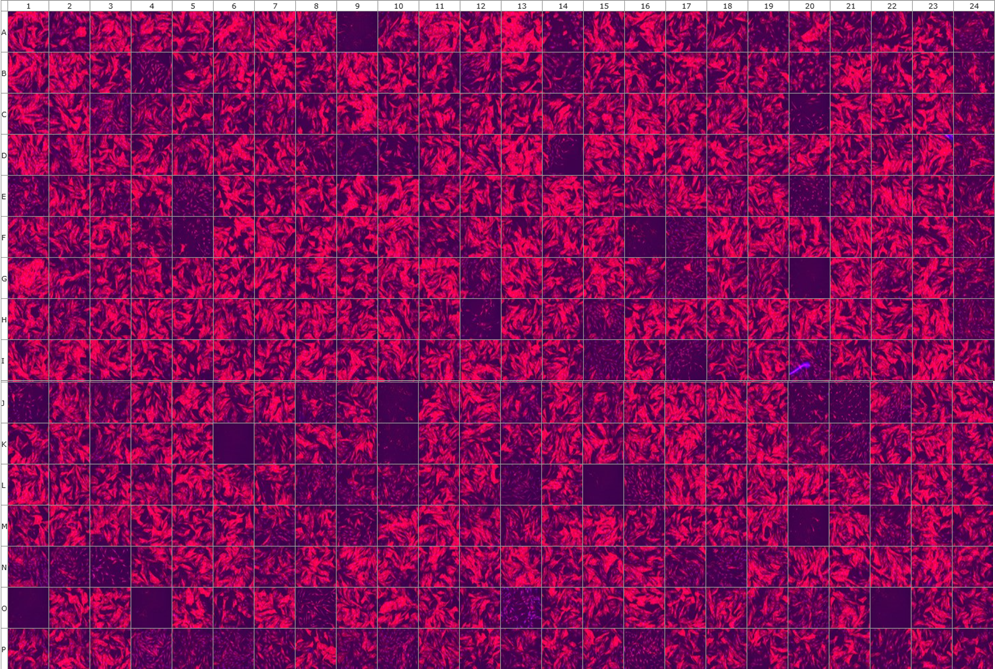
Representative picture of α-SMA stained myofibroblasts treated with 357 different compounds
References
- Hinz, B., et al., Alpha-smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol Biol Cell, 2003. 14(6): p. 2508-19.
- Jelaska, A., et al., Heterogeneity of collagen synthesis in normal and systemic sclerosis skin fibroblasts. Increased proportion of high collagen-producing cells in systemic sclerosis fibroblasts. Arthritis Rheum, 1996. 39(8): p. 1338-46.
- Lafyatis R. Transforming growth factor β-at the centre of systemic sclerosis. Nat Rev Rheumatol. 2014;1–14.
SSc is a heterogeneous disease with patient-to-patient variability that may lead to a wide variety of clinical manifestations. Patient subtypes also respond differently to therapies, hence, there is an unmet need for effective targeted therapies (1). Precision medicine, combining a deep analysis of biological and clinical data with bioinformatics to identify treatment responders, represent the most promising approach (2). However, it is unknown how to select available treatments and, in clinical practice, treatment strategies are following a trial and error principle.
In 2017, Stoeckius and colleagues developed a modified version of scRNA-seq allowing simultaneous cell phenotyping using oligonucleotide-labeled antibodies targeting surface markers and transcriptome measurements (CITE-seq) (3). CITE-seq allows the identification of rare, disease-relevant cell populations and the simultaneous characterization of transcriptomic changes. Indeed, specific cell subsets and molecular pathways could be linked with clinical phenotypes and response to therapy.
In this project, we aim to identify cellular and molecular biomarkers using CITE-seq that can predict, at baseline visit, disease activity and response to immunomodulatory treatments. To determine biomarkers for treatment response could allow the selection of the best treatment for each patient based on specific PBMC composition and activity.
References
- Gordon JK, Domsic RT. Clinical Trial Design Issues in Systemic Sclerosis: an Update. Curr Rheumatol Rep. 2016;18(6):38. doi:10.1007/s11926-016-0582-z
- Dobrota R, Mihai C, Distler O. Personalized Medicine in Systemic Sclerosis: Facts and Promises. Curr Rheumatol Rep. 2014;16(6):425. doi:10.1007/s11926-014-0425-8 doi: 10.1016/S2213-2600(20)30330-1. PMID: 33412120.
- Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865-868. doi:10.1038/nmeth.4380








