In einer Vielzahl von Analysemethoden werden weit über 50 verschiedene Autoantikörper gesucht. Auch spezialisierte Bereiche, wie z.B. neurologische Autoantikörper, werden mit einem grossen Spektrum abgedeckt. Notfallmässig bestimmen wir Autoantikörper im Rahmen von Abklärungen einer ANCA-assoziierter Kleingefässvaskulitis.
Autoantikörper bei Diabetes mellitus
Die Bestimmung der Antikörper gegen Glutamat Decarboxylase (GAD), Insulinoma Antigen 2 (IA2), Zinktransporter-8 (ZnT8) und Insulin erlaubt mit grosser Sensitivität die Entscheidung, ob ein Diabetes mellitus autoimmun bedingt ist oder nicht. Diese werden auch gemäss den internationalen ISPAD Consensus Guidelines empfohlen. Vor allem Antikörper gegen ZnT8 sind wichtig, da einige Patienten und Patientinnen nur Antikörper gegen diesen Zinktransporter aufweisen.
Antikörper gegen ZnT8 zusammen mit den Antikörpern gegen GAD65, IA2 und Insulin haben eine Sensitivität von 96% für einen autoimmunen Diabetes mellitus.
Diabetes und Antikörper
Diabetes mellitus Typ 1
Der Diabetes mellitus Typ 1 (DM1) ist eine chronische Erkrankung, bei der die Insulin produzierenden Beta-Zellen des endokrinen Pankreas zerstört werden. Die Krankheit verläuft dabei in Phasen: Autoimmunität, aber noch keine Symptome, abnormer Glukosetoleranztest, manifester DM1, Spätkomplikationen. Denn erst, wenn ca. 80-90% der Beta-Zellen zerstört sind, wird der DM1 manifest.
Beim Nachweis von Diabetes-Antikörper handelt es sich um einen Typ 1A (autoimmun), wenn diese fehlen um einen Typ 1B (idiopathisch). Ein DM1, der vor einem Alter von 6 Monaten auftritt, ist meistens durch eine Mutation im Transkriptionsfaktor FOXP3 bedingt und Teil eines IPEX-Syndroms (immunodysregulation polyendocrinopathy enteropathy X-linked syndrom).
LADA
LADA steht für Late onset autoimmune diabetes in the adult, also übersetzt: spät auftretender, autoimmun bedingter Diabetes beim Erwachsenen. Bei diesen Patienten und Patientinnen wird in der Regel zuerst ein Diabetes mellitus Typ 2 diagnostiziert. Unter den Typ 2-Diabetikern verstecken sich aber vermutlich etwa 10%, die an einem LADA leiden. Hinweise darauf ergeben sich aufgrund der folgenden Punkte: kein Übergewicht, Alter <50 Jahren, weitere Autoimmunerkrankungen beim Patienten, bei der Patientin oder in der Familie, niedrige C-Peptid- und Insulinspiegel, rascher Wirkverlust der oralen Antidiabetika und gutes Ansprechen auf Insulin. Der Nachweis von Diabetes-Antikörpern beweist dann das Vorliegen eines LADA.
Unterscheidung der verschiedenen Diabetes-Typen
Die Differenzierung zwischen Typ 1, Typ2, monogenen und anderen Diabetes-Formen hat Konsequenzen für die Therapie und Instruktion der Patienten und Patientinnen.
Zur Bestätigung eines Typ 1 Diabetes mellitus oder LADA stehen folgende AK zur Abklärung zur Verfügung. Im Gegensatz zu anderen Erkrankungen wird keine Stufendiagnostik empfehlen, sondern die gleichzeitige Bestimmung (ISPAD Consensus Guidelines). Bei Patienten und Patientinnen, die noch keinen Diabetes mellitus haben, steigt das Risiko an einem DM1 zu erkranken, mit der Anzahl der positiven Antikörper. Bei einem Antikörper ist das Risiko gering, bei mehreren hoch, so dass dann fast 100% nach 20 Jahren an einem DM1 erkrankt sind. Die Antikörper richten sich alle gegen Komponenten des sekretorischen Pathways.
|
(Glutamat Decarboxylase)
|
GAD65 ist ein peripheres Membranprotein der Mikrovesikel in den β-Zellen und verantwortlich für die Gamma-Aminobuttersäure-Synthese.
|
|
(Insulinoma Antigen, Tyrosin-phosphatase)
|
IA2 ist wichtig für die Sekretion des Insulins. Wir verwenden neu einen ELISA anstelle des RIAs. Wir verwenden die Version 1 des Testes mit dem Cut-off von <15 U/ml, nicht die aufwändigere Version 2 mit dem tieferen Cut-off von 7.5 U/ml. Vergleichsmessungen haben gezeigt, dass beide Versionen gleich sensitiv sind, da Patienten und Patientinnen mit einem autoimmunen Diabetes mellitus in der Regel hohe Werte für die Anti-IA2 im Bereich von 100-1000 U/ml aufweisen.
|
|
(Zinktransporter-8)
|
ZnT8 ist ein Kanal, durch den das Zink in die sekretorischen Granula der Insulin-produzierenden Zellen gelangt. Zink ist notwendig für die Kristallisierung der Insulin-Hexamere, die dense cores der Vesikel. Das Zink wird dann zusammen mit dem Insulin sezerniert.
Antikörper gegen ZnT8 werden bei 60-80% der Patienten und Patientinnen mit einem neu diagnostizierten Diabetes mellitus Typ I gefunden. Wichtig ist dabei, dass ein Viertel der Patienten und Patientinnen nur ZnT8-AK besitzen.
Diese Antikörper sind hauptsächlich gegen die intrazellulär gelegenen Aminosäuren 268-369 gerichtet. An Position 325 findet man einen Polymorphismus. Hier können die Aminosäuren Arginin (R) (75% bei Kaukasiern), Tryptophan (W) oder selten Glutamin (Q) vorkommen. Die Patienten und Patientinnen können entweder Antikörper nur gegen die von ihren Genen kodierte Variante oder gegen mehrere aufweisen. Antikörper gegen die Variante Q325 alleine sind sehr selten. Der von uns verwendete Assay von RSR enthält als gecoatetes Antigen ein Dimer der C-terminalen Domäne von ZnT8 (Aminosäuren 275-369) mit den Varianten R325 und W325. Gebundene Antikörper werden dann mit Biotin-markiertem ZnT8 nachgewiesen.
|
|
|
vor allem wichtig bei Kleinkindern, da in diesem Alter die Prävalenz hoch ist
|
|
|
Die Bestimmung wird in den internationalen ISPAD Guidelines 2014 nicht mehr empfohlen.
|
Ein oder mehrere dieser Antikörper sind bei 94% der Patienten und Patientinnen vorhanden, wenn das erste Mal eine erhöhte Nüchternglukose gemessen wird. Wenn jedoch keiner dieser Antikörper nachweisbar ist, dann sollte eine andere Form eines Diabetes mellitus in Betracht gezogen werden, falls einer der folgenden Punkte erfüllt ist:
• eine autosomal dominante Form eines Diabetes mellitus in der Familie
• Diagnose des Diabetes mellitus in den ersten sechs Lebensmonaten
• Nur leicht erhöhte, nicht progrediente Nüchternglukose (5.5-8.5 mmol/l) in jungen, gesunden, asymptomatischen Patienten und Patientinnen
• Assoziierte Symptome wie Taubheit, Optikusatrophie, Anzeichen eines Syndroms
• Einnahme von Substanzen, die bekanntermassen für Beta-Zellen toxisch sind oder Insulinresistenz auslösen können
Risikoabschätzung bei Patienten und Patientinnen, die noch nicht an einem Diabetes mellitus erkrankt sind
Wenn nur ein Antikörper nachweisbar ist, dann ist das Risiko für einen Typ 1-Diabetes mellitus in den nächsten 10 Jahren gering. Jährliche Kontrollen sind aber empfohlen, da Antikörper nur passager nachweisbar sein können oder weil neue dazukommen können. Bei Nachweis von mehreren Antikörpern ist das Risiko, in den nächsten Jahren zu erkranken, gross.
Autoantikörper bei Bullösen Autoimmundermatosen
Um den Körper vor schädlichen Einflüssen der Umwelt zu schützen und auch den mechanischen Belastungen standhalten zu können, müssen die Zellen der Haut durch stabile Kontakte zusammengehalten werden.
Desmosomen und Hemidesmosomen
In der Epidermis sind es die Desmosomen, die benachbarte Zellen mittels den transmembranären Proteinen Desmoglein 1/3 und Desmocollin, sowie den intrazellulären Plakinen zusammenhalten. Die basalen Zellen der Epidermis hingegen werden durch die Hemidesmosomen mit der darunterliegenden Basallamina verbunden. Hier sind es die intrazellulären Proteine bp230 und Plektin, die die Verbindung herstellen. Das bp230 bindet via bp180, das Plektin via Integrin α6β4 an das Laminin 332 (Laminin 5), das schlussendlich den Kontakt zum Kollagen Typ VII herstellt (siehe Abb. 1).
Entweder genetisch bedingt durch fehlende oder defekte Proteine oder erworben durch auto-reaktive T- und B-Lymphozyten kann die Funktion der Desmosomen und Hemidesmosomen gestört werden, so dass es zur Bildung von Blasen entweder in der Epidermis oder zwischen der Epidermis und der Dermis kommt.
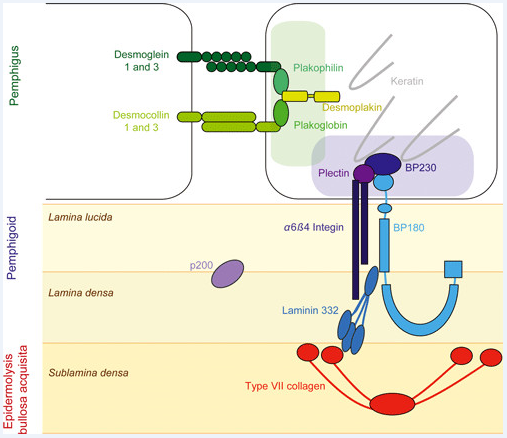
Abb. 1: Aufbau der Desmosomen und Hemidesmosomen. Die Autoantigene der Pemphiguserkranungen sind in grün, diejeniger der Pemphigoiderkrankungen in blau und das Hauptantigen der Epidermolysis bullosa acquisita ist in rot dargestellt (nach Bieber et al, 2010)
Intraepidermale Blasen: Pemphigusgruppe
Bei den Pemphiguserkrankungen führen Antikörper gegen Bestandteile der Desmosomen zu intraepidermalen Blasen der Haut und Schleimhäute. Die Antikörper sind pathogen. Dies zeigt sich daran, dass die Titer bei den meisten Patienten und Patientinnen mit der klinischen Aktivität korrelieren, dass Neugeborene von Müttern mit aktiver Erkrankung vorübergehend Blasen ausbilden und dass im Tierversuch die Injektion dieser Antikörper in neugeborene Mäuse zu Blasen führt.
Sowohl in der direkten als auch der indirekten Immunfluoreszenz (IF und iIF) lassen sich die Antikörper nachweisen. Für die indirekte Immunfluoreszenz wird Ösophagus verwendet. Es zeigt sich ein interzelluläres, wabenartiges Muster. Zielantigene sind vor allem Desmoglein 1 und 3, aber auch Desmocollin und Envoplakin und Periplakin.
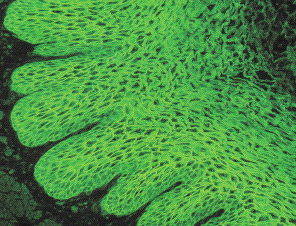
Abb. 2: iIF auf Ösophagus, Nachweis von Antikörpern gegen Desmosomen (dsg1 und dsg3)
Da dsg1 und dsg3 in der Haut und in der Schleimhaut unterschiedlich exprimiert werden, ergibt sich je nach Antikörper ein unterschiedlicher Phänotyp. Dsg1 wird vor allem an der Oberfläche der Epidermis exprimiert, aber nur wenig in Schleimhäuten, während dsg3 in den tieferen Schichten der Haut und in den Schleimhäuten lokalisiert ist (siehe Abb. 3).
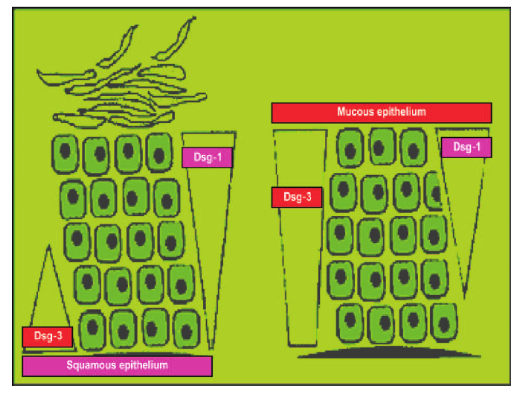
Abb. 3 Verteilung von dsg1 und dsg3 in der Haut links und in der Schleimhaut rechts (nach Tchernev et al, 2015)
Diagnostik bei intraepidermalen Blasen
Die Diagnostik erfolgt mittels indirekter Immunfluoreszenz auf Ösophagus und den ELISA zur Bestimmung der Antikörper gegen dsg1 und 3.
| Erkrankung |
Klinik |
Antikörper |
Diagnostik |
| Pemphigus vulgaris |
suprabasale Blasen der Haut und Mukosa |
|
- iIF auf Ösophagus
- Anti-dsg1/3 Sens. >90%
|
| suprabasale Blasen der Mukosa |
|
| Pemphigus foliaceus |
subkorneale Blasen, Mukosa nicht betroffen |
|
| Paraneoplastischer Pemphigus |
Hämorrhagische Stomatitis, Exantheme, Tumor (v.a. Non-Hodgkin-Lymphom) |
|
- iIF auf Rattenblase*
- Anti-Plakine
- Anti-dsg1/3
|
Subepidermale Blasen: Pemphigoid, Epidermolysis bullosa acquisita und Dermatitis herpetiformis Duhring
Bei den Pemphigoiderkrankungen führen Antikörper gegen Bestandteile der Hemidesmosomen zu supepidermalen Blasen. Die häufigste Pemphigoiderkrankung und auch die häufigste bullöse Autoimmundermatose ist das bullöse Pemphigoid. Es trifft meist ältere Patienten und Patientinnen. Da zu Beginn oft noch keine Blasen vorhanden sind, ist bei juckenden Hautveränderungen im Alter immer auch an ein bullöses Pemphigoid zu denken. Eine ähnliche Erkrankung ist das Pemphigoid gestationis, das selblimitierend meist im dritten Trimenon einer Schwangerschaft auftreten kann. Bei beiden Erkrankungen richten sich die Antikörper gegen bp180 (auch Kollagen XVII genannt) und bp230. Die Serumspiegel an Anti-bp180 korrelieren mit der Krankheitsaktivität. Die Antikörper richten sich meistens gegen die extrazellulär gelegene immundominante Domäne NC16A. Diese Domäne wird auch für die ELISAs verwendet.
Beim sehr seltenen Schleimhautpemphigoid kann es zu Vernarbungen kommen, die zu Blindheit oder Strikturen des Larynx oder Ösophagus führen können. Je nach Antikörper, wie im Falle von Antikörper gegen Laminin-332 kann die Erkrankung paraneoplastisch sein.
Die lineare IgA-Dermatose (LAD) ist sehr polymorph. Sie ist die häufigste bullöse Autoimmundermatose bei Kindern, kommt aber auch bei Erwachsenen vor. Es finden sich IgA-AK gegen LAD-1, ein Spaltprodukt von bp180.
Das Anti-p200-Pemphigoid, eine dem bullösen Pemphigoid-ähnliche Erkrankung ist durch Antikörper gegen ein p200 (Laminin-γ1-Kette) gekennzeichnet.
Die Epidermolysis bullosa acquisita ist eine schwere Erkrankung mit subepidermalen Blasen, welche durch mechanische Belastung der Haut entstehen und sekundär zu Erosionen der Haut führen. Die Antikörper richten sich vor allem gegen Konfirmationsepitope der NC-1-Domäne des Kollagen VII, können sich aber auch gegen andere Teile richten, was vielleicht die unterschiedlichen klinischen Manifestationen der Epidermolysis bullosa acquisita erklärt. Die Antikörper sind meist vom IgG-Isotyp, IgA kommt aber ebenfalls vor. Sie korrelieren mit der Krankheitsaktivität, sind also ein Aktivitätsmarker.
Die Dermatitis herpetiformis Duhring gilt als kutane Manifestation einer Zöliakie, wobei die Zöliakie selber meist mild verläuft. Die Patienten und Patientinnen leiden an einem heftigen Juckreiz und zeigen Papeln an den Streckseiten der Extremitäten und sakral.
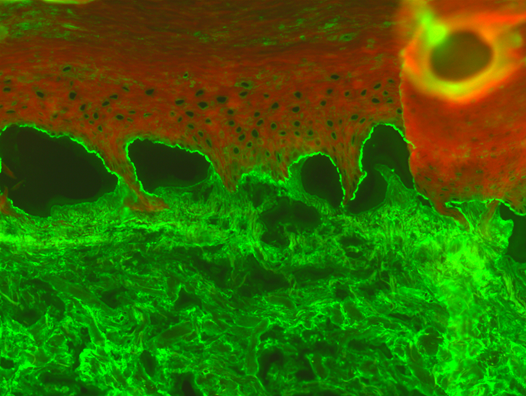
Abb. 4: indirekte Immunfluoreszenz auf Spalthaut, Nachweis von Antikörpern gegen die epidermale Seite (Anti-bp180, Anti-bp230)
Diagnostik bei subepidermalen Blasen
Die Diagnostik erfolgt mittels direkter sowie auch indirekter Immunfluoreszenz auf Ösophagus und auf Spalthaut. Zur Herstellung der Spalthaut werden Hautproben von Primaten mit 1 M NaCl inkubiert, wodurch sich ein Spalt in der Basalmembran bildet. Die Lokalisation der gebundenen Antikörper, am Blasendach oder am Blasenboden, lässt Rückschlüsse auf die Erkrankungen zu. Zudem stehen ELISAs zum Nachweis von Antikörpern gegen bp180 (NC16A) und bp230 zur Verfügung. Bei V.a. eine lineare IgA-Dermatose muss dies schriftlich vermerkt werden, damit die iIF mit einem IgA-Konjugat durchgeführt wird.
| Erkrankung |
Klinik |
Antikörper |
Diagnostik |
| Bullöses Pemphigoid (BP) |
Subepidermale Blasen vor allem der Haut, weniger der Mukosa, im Alter |
|
- iIF auf Spalthaut, epidermal,
Sensitivität: BP >90%, PG 30%
- Anti-bp180 und bp230
|
| Pemphigoid gestationis (PG) |
Meist drittes Trimenon, oft ohne Blasen |
|
| Schleimhaut-Pemphigoid |
Mukosa (Mund, Augen, Kehle), vernarbend |
- bp180
- Integrin α6β4
- Laminin 332
|
- IF mit linearer Ablagerung von IgG, IgA und Komplement C3 entlang der Basalmembran
- iIF auf Spalthaut, epidermal u/o dermal; Sensitivität 50%
- Anti-bp180
- Anti-Laminin-332
|
| Lineare IgA-Dermatose |
polymorph, Kinder |
|
- iIF auf Spalthaut, epidermal, IgA
|
| Anti-p200-Pemphigoid |
Subepidermale Blasen |
|
- iIF auf Spalthaut, dermal
|
| Epidermolysis bullosa acquisita |
Blasen der Haut und Schleimhaut, vernarbend |
- NC1-Domäne des Kollagen VII
|
- IF mit linearer Ablagerung von Immunglobulinen und/oder Komplement C3 an der dermoepidermalen Junktionszone
- iIF auf Spalthaut, dermal
- Anti-Kollagen VII
|
| Dermatitis herpetiformis Duhring |
Juckende Papeln, Zöliakie |
|
- Anti-Transglutaminase IgA
|
Autoantikörper bei Myasthenia gravis (MG)
Die MG ist klinisch durch eine abnorme Ermüdbarkeit der Skelettmuskulatur charakterisiert. Die Muskelschwäche nimmt jeweils im Verlaufe des Tages zu. Meist sind zuerst die Augenmuskeln befallen, was mit Doppelbildern und einer Ptosis einhergeht, wobei letztere zu einem müden Gesichtsausdruck führt.
- Anti-AChR: Die MG ist ein Paradebeispiel einer Antikörper-vermittelten autoimmunen Erkrankung. AChR-AK sind meist vorhanden und am Ort der Pathologie nachweisbar. Der AK-Transfer im Tierversuch oder in der Schwangerschaft löst die Erkrankung aus. Eine Immunisierung mit dem Target-Antigen führt zu den klinischen und elektrophysiologischen Veränderungen der MG. Thymusabnormitäten sind bei der MG häufig. Wie genau die humorale Autoimmunität induziert wird, ist aber noch unklar. Bei der MG liegt das Problem postsynaptisch. Die AChR-AK binden und führen zu einem Verlust der Rezeptoren, so dass das Endplattenpotential nicht mehr zur Auslösung der Muskelkontraktion ausreicht. Die fetalen Rezeptoren unterscheiden sich in einer Untereinheit von den adulten. Dadurch ist es möglich, dass Mütter, die nur AK gegen fetale Rezeptoren haben, selber kaum symptomatisch ist, das Kind intrauterin aber schwer betroffen ist. Man unterscheidet zwei Typen von AChR, die nikotinischen und die muskarinischen AChR. Die nitkotinischen AChR werden weiter unterteilt in die diejenigen der motorischen Endplatte (n1) und die neuronalen (n2), d.h. ganglionären. AK gegen n1 führen zu MG, AK gegen n2 zu autonomer Gangliopathie. Die Bestimmung der AK gegen die AChR der Endplatte erfolgt in unserem Labor mittels RIA. Der Test erfasst AK gegen fetale und adulte Rezeptoren.
- Anti-MuSK: AK gegen AChR sind bei 90% der Patienten und Patientinnen mit generalisierter und bei 50% mit okulärer MG vorhanden. Bei 25–50% der AChR-negativen MG-Patienten und -Patientinnen können AK gegen MuSK (muskelspezifische Rezeptor-Tyrosinkinase) vorhanden sein. MuSK ist sowohl embryonal für die
Bildung der Endplatte, wie auch später zum Erhalt der Endplatte wichtig. AK gegen MuSK sind mit einer speziellen Form der MG, die mehr die Schulter- und die Atemmuskulatur beeinträchtigt, assoziiert. Die Bestimmung erfolgt mittels ELISA.
- Anti-LRP4: Wenn weder AK gegen AChR noch gegen MuSK nachweisbar sind, können AK gegen LRP4, ein Molekül, das MuSK aktiviert, vorhanden sein. Die Bestimmung dieser AK bietet das Labor Volkmann in Karlsruhe an.
- Anti-Titin: Titin ist ein abundantes, sehr langes Molekül des Muskels. AK gegen Titin richten sich in der Regel gegen MGT-30, ein 30 kDa grosses Sequenzmotiv. Die Sensitivität ist mit 30% nicht hoch. Dennoch kann deren Nachweis von klinischer Bedeutung sein, indem der Nachweis bei Patienten und Patientinnen unter 60 Jahren mit einem Thymom assoziiert ist. Die Bestimmung erfolgt mittels ELISA.
Autoantikörper bei Lambert-Eaton-Myasthenie-Syndrom (LEMS)
Das LEMS ist ebenfalls wie die Myasthenis gravis charakterisiert durch eine abnorme Ermüdbarkeit der Muskulatur, hier ist aber mehr die proximale Beinmuskulatur betroffen, weniger die Muskulatur der Arme und der Augen. Autonome Störungen wie Mundtrockenheit oder orthostatische Dysregulation sind häufig. Typisch ist eine flüchtige Besserung der verminderten Kraft nach kurzer maximaler Willkürinnervation. In 2/3 der Fälle ist das LEMS paraneoplastisch, v.a. bei kleinzelligem Bronchuskarzinom. Diese Tumore sind neuroektodermalen Ursprungs und können neuronale Antigene exprimieren. Bei der Diagnose eines LEMS muss daher, falls nicht bereits bekannt, ein Tumor gesucht werden. Die Symptome eines LEMS können aber sehr stark den generalisierten Symptomen der Myasthenia gravis ähneln, so dass häufig das LEMS zunächst als Myasthenia gravis fehldiagnostiziert wird. Hier kann die Bestimmung der VGCC-Antikörper differentialdiagnostisch hilfreich sein.
- Anti-VGCC: Im Gegensatz zur postsynaptischen Problematik bei der MG, liegt das Autoantigen beim LEMS präsynaptisch. Durch AK gegen die spannungsabhängigen Kalziumkanäle (vor allem vom P/Q-Typ) unterbleibt der Kalziumeinstrom, der für die Exozytose der mit Acetylcholin gefüllten Vesikel notwendig ist. Die Sensitivität dieser AK ist sehr gut, die Spezifität ebenfalls. Sie sind aber
auch positiv bei Kleinhirndegeneration im Rahmen eines kleinzelligen Bronchialkarzinom, ohne dass ein LEMS vorliegen muss. AK gegen VGCC bestimmen wir mittels eines RIA.
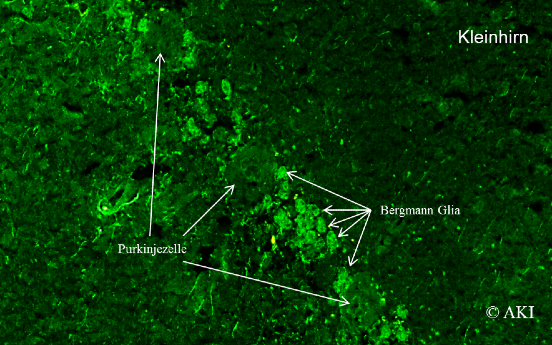
- Anti-SOX1: Anti-SOX1, auch Anti-Glia-nukleäre AK (AGNA) genannt, richten sich gegen ein Protein in den Zellkernen der Bergmann-Glia in der Purkinje-Zelllschicht des Kleinhirns. Bei Patienten und Patientinnen mit idiopathischen LEMS sind diese AK nicht zu finden, bei paraneoplastischem LEMS bei 2/3 der Patienten und Patientinnen. Wir bieten die Bestimmung der SOX1-AK in unserer Analyse Anti-ZNS, AK gegen onkoneuronale AK an. Im positiven Fall kontrollieren wir das Resultat mittels indirekter Immunfluoreszenz.
Autoantikörper bei Neuromyelitis optica (NMO)
Die Neuromyelitis optica (NMO, Devic-Syndrom) wurde lange als eine klinische Variante der Multiplen Sklerose angesehen. Spätestens seit 2004 die Antikörper gegen Aquaporin4 (Anti-AQP4 oder Anti-NMP) geschrieben wurden, ist klar, dass es sich um eine andere Erkrankung mit schlechterer Prognose handelt, die therapeutisch ganz anders angegangen werden muss. Klinisch präsentiert sich die NMO meistens mit einer ein- oder beidseitigen Optikusneuritis und/oder einer longitudinalen transversen Myelitis, die unbehandelt eine schlechte Prognose zeigen, d.h. ein Visusverlust oder eine Querschnittslähmung können die Folge sein. Zusätzliche Symptome wie Hirnstammenzephalitis mit Schluckauf, Erbrechen, Narkolepsie und neuroendokrinen Störungen sind auch möglich. Vor allem in Kindern sind auch weitere enzephalitische Manifestationen beschrieben.
Weil die betroffenen Strukturen nicht geeignet für eine Biopsie sind, stützt sich die Diagnose vor allem auf die Klinik, die Bildgebung und die Serologie. Bei Verdacht auf NMO können folgende Antikörper Anti-Aquaporin 4 und Anti-MOG bestimmt werden. Im negativen Fall wäre dann auch die Bestimmung von ANA, ANCA und der paraneoplastischen Antikörper sinnvoll, vor allem Anti-Hu und Anti-CV2/CRMP5. Der Liquor ist in einem Drittel der Patienten und Patientinnen normal, bei den anderen findet man eine lymphozytäre Pleozytose, in einem Drittel eine Proteinerhöhung und oligoklonale Banden nur in 10-30%.
- Antikörper gegen Aquaporin 4 (Anti-AQP4, Anti-NMO): Sie richten sich gegen Aquaporin 4. Dies ist der häufigste Wasserkanal im ZNS, Er wird reichlich auf den Astrozyten, an den Fortsätzen an der Blut-Hirn-Schranke exprimiert. Histologisch findet man eine Schädigung der Astrozyten mit Ablagerung von Immunglobulin und Komplement. Die Antikörper bleiben meistens nachweisbar, auch wenn der Patient oder die Patientin sich in Remission befindet.
- Antikörper gegen MOG (Anti-MOG): Diese Antikörper richten sich gegen das Myelin Oligodendrocyte Glycoprotein. Sie wurden bei einer Untergruppe von Patienten und Patientinnen mit NMO ohne Nachweis von Antikörpern gegen Aquaporin 4 gefunden. Wenn Anti-MOG nachgewiesen werden kann, ist die Prognose vermutlich besser. Auch histologisch findet man Unterschiede. In der Remission verschwinden die AK bei der Hälfte der Patienten und Patientinnen.
Autoantikörper bei Enzephalitiden
Bei Patienten und Patientinnen mit psychiatrischen Symptomen, vor allem wenn kombiniert mit neu aufgetretenen epileptischen Anfällen oder neurologischen Ausfällen, muss nicht nur eine Erreger-bedingte Ursache ausgeschlossen werden, sondern es muss auch an eine autoimmune Genese gedacht werden. Solche Erkrankungen sind vermutlich viel häufiger als man früher dachte. In den letzten Jahren sind viele Erkrankungen bekannt geworden, die durch Antikörper gegen Oberflächenantigen, Rezeptoren oder Kanäle, ausgelöst werden. Die Antikörper lösen die Symptome der Erkrankungen direkt aus, da sie zu einem Funktionsverlust der Antigene, d.h. der Rezeptoren und Kanäle führen. Eine frühzeitige Diagnose ist wichtig, da bei rascher Immuntherapie die Prognose besser ist, also weniger neurologische Defizite bleiben werden. Die Erkrankungen sind oft nicht paraneoplastisch, treten also meist nicht im Rahmen einer Tumorerkrankung auf. In der Regel ist der Verlauf monophasisch, Rezidive können aber, vor allem je nach Antikörper, auftreten. Nicht nur der Mensch kann solche Antikörper entwickeln, auch andere Säugetiere können erkranken. Knut, der Eisbär, ist aufgrund eines Anfalls ausgelöst durch eine NMDA-Rezeptor-Enzephalitis ertrunken.
In den nächsten Jahren werden sicher weitere Antikörper dazukommen. Wichtig ist es, daran zu denken und die Antikörper zu suchen, damit die Diagnose rasch gestellt und der Patient oder die Patientin adäquat behandelt werden kann.
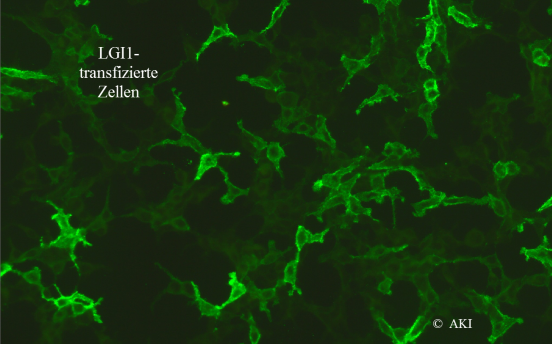
Diagnostik: Der Liquor kann entzündlich verändert sein (lymphozytäre Pleozytose, Proteinerhöhung, leichte Erhöhung des Albuminquotienten als Ausdruck einer Schrankenstörung und ev. oligoklonale Banden. Im MRI kann eine Entzündung des Hippocampus zu sehen sind. Da Liquor und MRI aber auch normal sein können, ist der Nachweis der Antikörper im Serum und eventuell auch im Liquor besonders wichtig. Die Detektion dieser Antikörper erfolgt mittels indirekter Immunfluoreszenz auf transfizierten Zellen (siehe Abb.). Denaturierende Methoden wie Immunoblots sind nicht geeignet.
Da sich die Symptome der einzelnen Erkrankungen überlappen, ist es sinnvoll, einen Screen mit den wichtigsten Antikörpern durchzuführen. Wir bieten Ihnen folgenden Enzephalitis-Screen an: AK gegen LGI1, CASPR2, NMDA-Rezeptor, GABAB-Rezeptor, AMPA1/2-Rezeptor, DPPX. Als Material reicht in der Regel Serum. Bei negativem Resultat und bestehendem Verdacht kann auch Liquor untersucht werden. Zusätzlich bieten wir Ihnen auch die Bestimmung der Antikörper gegen metabotropen Glutamatrezeptor 5 und Glycin-Rezeptor an.
NMDA-Rezeptor-AK-Enzephalitis
Glutamat ist ein erregender Neurotransmitter. Es gibt zahlreiche Glutamat-Rezeptoren. Ein besonders wichtiger heisst NMDA-Rezeptor. Der Name kommt daher, dass dieser Rezeptor durch das synthetische Molekül NMDA aktiviert wird. Enzephalitiden ausgelöst durch Antikörper gegen den NMDA-Rezeptor gehören zu den häufigeren autoimmunen Enzephalitiden. Diese Erkrankung wurde ursprünglich von Dalmau bei jungen Frauen oder Mädchen mit einem Teratom des Ovars beschrieben. Es hat sich aber gezeigt, dass auch andere Altersgruppen und Männer betroffen sein können und oft kein Teratom oder Tumor gefunden werden kann. Die Erkrankungen kann auch durch eine Herpes-Enzephalitis getriggert werden. Der Verlauf ist mehrphasig. Sie beginnt mit psychiatrischen Symptomen, was oft zur Aufnahme in ein psychiatrisches Krankenhaus führt. Dann kommen kognitive Einbussen und epileptische Anfälle hinzu. Unbehandelt kann die Erkrankung fortschreiten. Lähmungen und Bewusstseinsstörungen kommen dazu. Gefährlich ist die autonome Dysregulation, die einer intensivmedizinische Betreuung bedarf. Während der Genesung bilden sich dann die Symptome wieder zurück. Defizite können aber bestehen bleiben.
Enzephalitis durch Antikörper gegen AMPA1 und 2-Rezeptoren
Neben den NMDA-Rezeptoren gibt es noch weitere Glutamat-Rezeptoren. Die Glutamat-Rezeptoren 1 und 2 werden nach dem synthetischen Agonisten auch AMPA-Rezeptoren genannt. Es sind ionotrope Rezeptoren. AMPA-Rezeptoren sind wichtig für die synaptische Plastizität, z.B. im Hippocampus. An einer Enzephalitis ausgelöst durch die Antikörper erkranken vor allem ältere Frauen. Es beginnt mit Verwirrung, dann kommen Gedächtnisstörungen, eventuell kombiniert mit limbischen Symptomen und epileptischen Anfällen dazu. Der Verlauf kann fulminant mit Fieber, Koma und Hypertonie sein. Der Liquor kann eine Pleozytose und Proteinerhöhung zeigen. Das MRI zeigt meistens Läsionen, je schwerer diese sind, desto schlechter scheint die Prognose zu sein. In 70% der Fälle steckt ein Tumor (Thymom, Mamma- oder Bronchialkarzinom) hinter der Erkrankung. Die neurologischen Symptome sprechen gut auf eine Immunotherapie an, Rezidive sind aber häufig.
Die Bestimmung der Antikörper gegen AMPA-Rezeptoren erfolgt mittels indirekter Immunfluoreszenz auf transfizierten Zellen. In einem ersten Schritt erfolgt ein Screening auf Antikörper gegen AMPA1- und AMPA2-Rezeptoren. Nur im positiven Fall wird dann die Einzeltestung durchgeführt.
Enzephalitis durch Antikörper gegen den GABAB-Rezeptor
Wenn GABA, ein wichtiger hemmender Neurotransmittor, an den GABAB-Rezeptor bindet, dann führt dies prä- und postsynaptisch zu einer Hemmung, d.h. die Ausschüttung von Transmittern wird vermindert und die Auslösung eines Aktionspotentials wird erschwert, was eine exzessive neuronale Synchronisation verhindert. Bei Patienten und Patientinnen mit einer Enzephalitis ausgelöst durch Antikörper gegen den GABAB-Rezeptor sind diese Hemmung gestört. Sie präsentieren sich mit einer limbischen Enzephalitis mit Desorientierung, Verhaltensänderungen, Halluzinationen, Schlafstörungen und Temporallappen-Epilepsie. Der Liquor zeigt eine lymphozytäre Pleozytose und im MRI sind meist Veränderungen im medialen Temporallaben zu sehen. Die GABAB-Rezeptor-Antikörper können mit Antikörpern gegen GAD und SOX1 assoziiert sein. Die Hälfte der Patienten und Patientinnen hat eine paraneoplastische Erkrankung im Rahmen eines kleinzelligen Bronchuskarzinoms. Die neurologischen Symptome sollen gut auf eine Immuntherapie ansprechen.
Erkrankungen ausgelöst durch Antikörper gegen GAD
Das Enzym Glutamat-Decarboxylase (GAD), das den wichtigsten aktivierenden Neurotransmitter, in Gamma-Aminobuttersäure (GABA), den wichtigsten inhibierenden Neurotransmittor umwandelt, ist weder ein Rezeptor noch ein Ionenkanal. Es ist ein intrazelluläres Molekül: Da es aber an der Synapse zusammen mit den Neurotransmittoren freigesetzt wird, kann es von Autoantikörpern erkannt werden. Das Enzym wird im Nervensystem und in der Bauchspeicheldrüse exprimiert. Dies erklärt, dass Autoantikörper gegen GAD einerseits beim Diabetes mellitus Typ I, andererseits bei neurologischen Erkrankungen gefunden werden können. Beim Diabetes mellitus sind die Titer niedrig, bei den neurologischen Erkrankungen hingegen sehr hoch. GAD-AK können verschiedenste Erkrankungen im Nervensystem auslösen. Beschrieben sind zerebelläre Ataxie, limbische Enzephalitis, Stiff-Person-Syndrom (SPS) und die progressive Enzephalomyelopathie mit Rigidität und Myoklonus (PERM). Das SPS ist eine seltene Erkrankung, die assoziiert mit zwei verschiedenen Antikörper auftreten kann. Beim Nachweis von Anti-GAD ist meist kein Tumor zu finden, wenn aber Antikörper gegen Amphiphysin positiv sind, ist die Erkrankung paraneoplastisch im Rahmen eines Mammakarzinoms. Ein SPS wird anfänglich oft als psychiatrische Erkrankungen missgedeutet. Wegen einer Tonuserhöhung der Muskulatur treten Krämpfe auf, die durch Geräusche, Berührungen oder Emotionen getriggert werden.
Enzephalitis durch Antikörper gegen LGI1 und CASPR2
Spannungsabhängige Kaliumkanäle (VGKC) gibt es im ganzen Gehirn und dienen der Wiederherstellung des Membranpotentials während der Hyperpolarisation. Antikörper gegen LGI1 und CAPR2 richten sich dabei gegen Proteine, die mit dem VGKC assoziiert sind. Patienten und Patientinnen mit diesen Antikörpern sind oft männlich und leiden an limbischer Enzephalitis mit extralimbischen Symptomen wie Neuromyotonie mit schmerzhaften Muskelkrämpfen, Bewegungsstörungen, Schlafstörungen und Serumhyponatriämie. Die Häufigkeit dieser Symptome hängt vom nachgewiesenen Antikörper ab. Typisch für LGI1-Antikörper sind die faziobrachialen dystonen Anfälle. Es handelt sich um einseitige, ganz kurze Anfälle mit Dystonie des Armes und Grimassieren. Da sie bis zu Hunderten von Malen täglich auftreten können, stören sie die Patienten stark. Ausgelöst werden diese Anfälle durch Bewegungen, Emotionen und laute Geräusche. Sie treten vor der limbischen Enzephalitis auf, erlauben so eine frühzeitige Diagnose und Therapie, was bleibende kognitive Defizite vermeiden kann. Bei Anti-CASPR2-positive Patienten und Patientinnen muss man auf eine Dysautonomie achten, da diese Patienten und Patientinnen eine Bradykardie und plötzlichem Herztod erleiden können.
Antikörper die sich direkt gegen den VGKC richten sind nicht pathogen und werden bei Patienten und Patientinnen mit nicht autoimmunen Erkrankungen wie Morbus Alzheimer oder Parkinson gefunden. Aber auch bei Gesunden, vor allem bei gesunden Metzgern, die mit den Aerosolen von Tiergehirnen in Kontakt kommen werden sie gefunden. Aus diesem Grund wird die direkte Bestimmung der Anti-VGKC nicht mehr empfohlen.
Enzephalitis durch Antikörper gegen DPPX
DPPX ist wichtig für die Funktion des Kaliumkanals Kv4, der wiederum eine wichtige Rolle bei der synaptischen Plastizität spielt. DPPX kommt nicht nur im ZNS, hauptsächlich im Cerebellum und Hippocampus vor, sondern wird auch im Plexus myentericus exprimiert, was die gastrointestinalen Symptome erklärt. Klinisch zeigt sich meistens zuerst eine Diarrhoe, die von einer rasch progressiven Enzephalopathie mit Agitation, Halluzinationen, epileptischen Anfällen und Myoklonus gefolgt wird. Der Liquor zeigt Pleozytose, intrathekale IgG-Produktion und/oder oligoklonale Banden.
Enzephalitis durch Antikörper gegen GlyR
Der Glycin-Rezeptor kommt vor allem im Hirnstamm und Rückenmark vor, daneben auch noch in Spermien und Makrophagen. Es ist ein Ionenkanal. Wenn Glycin bindet, gelangen Chlorid-Ionen in die Zelle. Glycin ist neben dem GABA der wichtigste inhibitorische Neurotransmittor. Durch die Antikörper wird diese Hemmung behindert, die Muskeln reagieren zu stark, es kommt zur PERM (progressive Enzephalitis mit Rigidität und Myoklonus), der Maximalform eines Stiff-Person-Syndroms. Eine Tumorassoziation ist möglich. Das Ansprechen auf Immunotherapien ist ziemlich gut.
Enzephalitis durch Antikörper gegen mGluR5
mGluR5 ist ein G-Protein-gekoppelter Rezeptor und an der synaptischen Plastizität beteiligt. Er kommt vor allem im Hippocampus und in der Amygdala vor. Antikörper kommen auch bei jungen Personen vor und führen zu psychiatrischen Symptomen wie Agitation, Desorientierung, Halluzinationen und anterogradem Gedächtnisverlust. Das ursprünglich beschriebene Ophelia-Syndrom, benannt nach der Geliebten Hamlets, war eine limbische Enzephalitis mit neuropsychiatrischen Symptomen bei Hodgkin-Lymphom Das Lymphom ist aber nicht obligat. Das Syndrom spricht gut auf Steroide an.
Autoantikörper gegen MAG bei Polyneuropathie
Myelin-assoziiertes Gliykoprotein
Das Myelin-assoziierte Glykoprotein (MAG) ist ein 100 kDa transmembranes Protein, welches sowohl im ZNS als auch im peripheren Nervensystem (PNS) exprimiert wird. Es ist u.a. notwendig für die Formation der intakten Myelinlamellen durch die Oligodendrozyten und Schwann’schen Zellen. Obwohl die Konzentration im ZNS höher ist als im PNS, führen Antikörper gegen MAG nur zu einer peripheren Pathologie, da die intakte Hirnschranke den Übertritt von MAG-Antikörpern und Komplement ins ZNS verhindert.
Manifestationen bei Anti-MAG-Polyneuropathie
Ausser Diabetes mellitus und Alkohol sind monoklonale Gammopathien eine wichtige Ursache der Neuropathie. Meistens handelt es sich um ein monoklonales IgM und oft sind Anti-MAG-Antikörper nachweisbar. Als ursächliche Erkrankung ist ein MGUS (monoklonale Gammopathie unklarer Signifikanz) am häufigsten, gefolgt vom Morbus Waldenström, Non-Hodgin-Lymphom und chronischer Leukämie. Je nach Höhe der Anti-MAG-Titer handelt es sich vermutlich um andere Erkrankungen mit anderer Pathogenese.
- Hohe Titer >8000 E/ml bei klassischer Anti-MAG-Polyneuropathie
Bei hohen Titern der Anti-MAG-Antikörpern (>8000 E/ml) kommt es zur klassischen Anti-MAG-Polyneuropathie. Diese ist durch eine symmetrische, distal betonte, langsam progrediente sensorisch-ataktische Neuropathie charakterisiert. Neuropathische Schmerzen können auch auftreten. Betroffen sind vor allem ältere Personen und Männer häufiger als Frauen.
In der Elektroneurographie zeigt sich eine demyelinisierende Neuropathie. In der Nervenbiopsie sieht man eine Abnahme der myelinisierten Fasern und lymphozytäre Infiltrate. Im EM sind bei einem Teil der noch verbleibenden Fasern im Elektronenmikroskop vergrösserte Abständen zwischen den äusseren Lamellen der Markscheide nachweisbar. Verursacht wird diese Auflockerung der Lamellen durch die Ablagerung der monoklonalen Antikörper.
Mittels indirekter Immunfluoreszenz auf Nerven-Substrat mit einem IgM-Konjugat findet man Antikörper gegen Nerv.
- Tiefe Titer <8000 E/ml bei CIDP-ähnlicher Erkrankung
Bei hohen Titern der Anti-MAG-Antikörper (<8000 E/ml) kommt es zu einer CIDP-ähnlichen Erkrankung. CIDP steht für chronisch inflammatorische demyelinisierende Polyneuropathie. Diese Patienten und Patientinnen haben eine sensorisch-motorische, demyelinisierende Neuropathie. In der histologischen Untersuchung finden sich keine vergrösserten Abstände der Lamellen und die indirekte Immunfluoreszenz ist negativ.
Onkoneuronale Autoantikörper bei paraneoplastischen neurologischen Syndromen
Onkoneuronale Antikörper finden sich bei paraneoplastischen neurologischen Syndromen (PNS). Man geht davon aus, dass es im Rahmen einer anti-tumoralen Immunreaktion zur Bildung dieser Antikörper kommt. Die Autoantikörper richten sich dabei aber nicht nur gegen Tumorgewebe, sondern auch gegen Strukturen des Nervensystem. Neurologischen Symptome treten daher zwar unmittelbar im Zusammenhang mit dem Tumorleiden auf, sind jedoch nicht direkt durch den Tumor oder seine Metastasen verursacht. Die Symptome sind sehr vielfältig, daher sollte ein paraneoplastisches Syndrom frühzeitig in die differentialdiagnostischen Überlegungen einbezogen werden. Besonders häufig präsentieren sich die Patienten und Patientinnen mit Zeichen einer limbische Enzephalitis oder einer Kleinhirndegeneration. Die Suche nach onkoneuronalen Antikörpern ist zudem von besonderer klinischer Bedeutung, da die neurologischen Symptome der Entdeckung des auslösenden Tumors in der Regel Monate bis Jahre vorausgehen. Ausserdem gibt es typische Assoziationen gewisser Autoantikörper mit bestimmten Tumoren und neurologischen Symptomen. Die Diagnostik kann hier also einen entscheidenden Beitrag zur Früherkennung von Krebserkrankungen leisten.
| Antikörper |
Paraneoplastisches Syndrom |
Assoziierte Tumore |
|
|
Sensible und/oder autonome Neuropathie
Kleinhirndegeneration (Ataxie)
Enzephalomyelitis
Limbische Encephalitis
|
kleinzelliges Bronchialkarzinom, grosszelliges Bronchialkarzinom, extrapulmonale kleinzellige Karzinome, u.a.
|
|
|
Hirnstamm Syndrom (inkl. Opsoklonus-Myoklonus-Syndrom)
Kleinhirndegeneration
|
Mammakarzinom, Bronchialkarzinom, Medulläres Schilddrüsenkarzinom, u.a.
|
|
|
paraneoplastische Kleinhirndegeneration
|
|
|
|
Limbische Enzephalitis
Hirnstamm Syndrom
Kleinhirndegeneration
|
Testikuläre Tumore (oft junge Patienten und Patientinnen)
|
|
|
Stiff-Person-Syndrom
Limbische Enzephalitis
|
Sowohl paraneoplastisch als auch nicht paraneoplastisch vorkommend
|
|
|
|
Mammakarzinom, (Kleinzelliges Bronchialkarzinom)
|
|
|
Kleinhirndegeneration (Ataxie)
|
Ovarialkarzinom, Mammakarzinom, Endometriumkarzinom, u.a.
|
|
|
|
|
|
|
Lambert Eaton Myasthenia Gravis
|
Kleinzelliges Bronchialkarzinom
|
|
Anti-CV2
|
Sensible und sensomotorische Neuropathie
Enzephalomyelitis
Kleinhirndegeneration
Limbische Enzephalitis
Autonome Neuropathie
Chorea
|
Kleinzelliges Bronchialkarzinom, Thymom
|
|
Anti-ZIC4
|
|
Kleinzelliges Bronchialkarzinom
|
adaptiert von: Wandinger KP, MTA Dialog 4, 2008
In unserem Diagnostiklabor verwenden wir zur Detektion dieser Autoantikörper einerseits einen Immunodot-Assay, anderseits die indirekte Immunfluoreszenz. Für letztere werden die Patientenseren mit Präparaten von Kleinhirnschnitten inkubiert. Befinden sich im Patientenserum Antikörper, welche an dieses Präparat binden, werden sie durch anschliessende Inkubation mit fluoreszierenden Antikörpern für die Analyse mit dem Fluoreszenzmikroskop sichtbar gemacht. Bei Patienten und Patientinnen, die aufgrund einer rheumatologischen Erkrankung anti-nukleäre Autoantikörper (ANA) haben, können diese ebenfalls eine positive Fluoreszenz der Zellkerne auf Cerebellum-Präparaten zeigen – in diesen Fällen ist die indirekte Immunfluoreszenz auf Cerebellum-Substrat nicht beurteilbar.
Schlafstörungen durch Autoantikörper gegen IgLON5
IgLON5-Antikörper sind bis jetzt vor allem bei älteren Personen beschrieben worden (ab der 5. Dekade). Die Erkrankung weist sowohl autoimmune als auch neurodegenerative Charakteristika auf. Histologisch findet man eine Tauopathie im Hypothalamus und im Tegmentum des Hirnstamms. Das MRI des Gehirns, EEG und der Liquor sind meistens unauffällig.
Klinisch ist die Erkrankung heterogen. Typisch sind Schlafstörungen, bulbäre Symptomen (Hirnstamm), Gangstörungen, Probleme des autonomen Nervensystems und nicht selten auch eine Demenz. Die häufigsten Symptome finden Sie in untenstehender Tabelle. Da diese Symptome bei älteren Personen auch wegen anderen Ursachen oft auftreten, ist die Erkennung des Krankheitsbildes nicht einfach.
Häufigste Symptome assoziiert mit IglON5-Antikörpern
- Schlafstörungen
- Bulbäre Symptome
- Ganginstabilität
- Dysautonomie
- Kognitive Dysfunktion
- Schlafapnoe, Parasomnie, Insomnie, Tagesmüdigkeit
- Schluckstörung, Sprechprobleme, vermehrter Speichelfluss
- Stürze, Schwindelgefühl
- Harninkontinenz, Schweissausbrüche, Herzrhythmusstörungen
- Demenz
Abklärung bei membranöser Glomerulonephritis
Die membranöse Glomerulonephritis (MGN) ist eine chronisch entzündliche Erkrankung der Glomeruli in der Niere. Sie ist im Erwachsenenalter die häufigste Ursache des nephrotischen Syndroms. Beim nephrotischen Syndrom gehen viele Proteine über den Urin verloren, was u.a. zu Ödemen führt. Mikroskopisch findet man eine Verdickung der Basalmembran in den Glomeruli, die infolge der Ablagerung von Proteinen auftritt. Die MGN kann sekundär im Rahmen anderer Erkrankungen auftreten, z.B. bei einem SLE oder einer Hepatitis B-Infektion, sie kann aber auch sogenannt idiopathisch auftreten. Idiopathisch bedeutet «ohne erkennbare Ursache», aber auch «unabhängig von anderen Krankheiten». Denn 2009 beschrieben Beck et al. m Serum bei 70% der Patienten mit idiopathischer MGN Antikörper gegen des PLA2-Rezeptor, ein Protein auf den Podozyten in den Glomeruli. Bei einigen der restlichen Patienten kann ein weiterer Antikörper, nämlich gegen den THSD7A, nachgewiesen werden. Die Ursache ist somit nicht mehr gänzlich unklar, es handelt sich um eine direkte Schädigung durch die Ablagerung von Autoantikörpern.
Testung auf Anti-PLA2R-Antikörper:
Die KDIGO-Leitlinien führen die Bestimmung mittels ELISA als Test für das Monitoring an. Mit dem von der Firma angegebenen Referenzwert des ELISAs von <14 RE/ml erhielten wir eine Sensitivität von nur 60%. Aus diesem Grunde verzichteten wir anfänglich auf die Einführung des ELISAs. Anhand von 60 positiven Patientenproben haben wir den Test dann reevaluiert und einen eigenen Referenzwert bestimmt. Mit dem neuen Referenzwert von <2 RE/ml konnten wir bei weiterhin sehr guter Spezifität die Sensitivität auf 98% steigern. Dies passt mit den Daten von Bobart et al. überein.
Wir können Ihnen nun folgendes Algorithmus vorschlagen.
- Screening mittels iIF und ELISA für eine optimale Sensitivität und um einen Ausgangswert für den ELISA zu etablieren
- Monitoring mittels ELISA
Im negativen Fall empfiehl sich dann als weitere Abklärung die Bestimmung des Anti-THSD7A-Antikörper.
Weitergehende Abklärungen bei unklaren ANCA-Mustern
Für die Abklärung von unklaren ANCA-Mustern, d.h. deutlich positive ANCA in der iIF, aber negative oder nur unwesentlich positive MPO- oder PR3-Antikörper, können wir Ihnen nun eine deutlich erweiterte Palette an Antikörpern anbieten
Autoantikörper gegen BPI:
BPI (Bactericidal Permeability-Increasing Protein) ist ein wichtiges endogenes Protein in der Abwehr von Gram-negativen Bakterien. Antikörper gegen BPI werden vor allem bei Patienten mit zystischer Fibrose, Bronchiektasen, chronisch entzündlichen Darmerkrankungen wie Morbus Crohn oder Colitis ulcerosa, reaktiver Arthritis und primär sklerosierender Cholangitis gefunden. Sie verschlechtern die Abwehr des Patienten und führen zu schwereren Lungenschäden bei Lungenentzündungen. Bei Patienten mit zystischer Fibrose sind diese Antikörper prognostisch ungünstig, weil sie die Patienten anfällig für Infektion mit P. aeruginosa machen.
iIF: zytoplasmatisches Muster, die Antikörper gegen PR3 sind aber negativ
Autoantikörper gegen Elastase:
Elastase ist eine vor allem in polymorphkernigen neutrophilen Granulozyten, vorkommende Serinprotease, die wichtig für die Immunabwehr ist. Ferner ist Elastase massgeblich an der Gewebezerstörung bei Emphysem und rheumatoider Arthritis beteiligt. AK gegen die Elastase kommen vor allem bei Kokain-Abusus vor und werden durch das Streckmittel Levamisol induziert. Klinisch manifestiert sich die Erkrankung meist als eine Kokain-induzierte destruktive Mittellinienläsion oder ein Kokain-assoziiertes Autoimmunsyndrom, mit Arthralgien und Hautläsionen. Diese Erkrankung kann aber auch durch Medikamente ausgelöst werden, z.B. durch Propylthiouracil, Hydralazin und Minocyclin.
iIF: p-ANCA mit betontem Rand, atypisch p-ANCA
Autoantikörper gegen Kathepsin G:
Kathepsin G kann die Umwandlung von Angiotensin I in Angiotensin II katalysieren, sowie Bindegewebsproteine (z.B. Kollagene, Fibronektin) abbauen. Anti-Kathepsin G Antikörper wurden bei Colitis ulcerosa (40%), SLE, RA und Morbus Crohn nachgewiesen. Der Titer korreliert mit der Aktivität der Colitis ulcerosa.
iIF: p-ANCA
Autoantikörper gegen Laktoferrin (LF):
LF kommt in den sekundären Granula der Granulozyten und in der Tränenflüssigkeit und in der Muttermilch vor. Es bindet Eisen und wirkt dadurch bakteriostatisch, da die meisten pathogenen Bakterien Eisen zum Wachstum benötigen. Zudem hat LF anti-inflammatorische Funktionen. Es reduziert die Menge an IL6, TNF-alpha, IL8 und hemmt die Ablagerung von C3 auf Immunkomplexen. AK gegen LF könnten also die Entzündungsreaktion im Körper fördern. AK gegen LF werden z. B. bei Patienten mit chronisch entzündlichen Darmerkrankungen, bei rheumatoider Arthritis mit Vaskulitis, AIH und SLE gefunden. Beim SLE korrelieren hohe Titer mit erhöhter Krankheitsaktivität.
iIF: p-ANCA